
For generations, biologists have aimed to understand every single process of life. The key to moving closer to this goal is to further understand the compounds that govern almost all life processes, i.e., proteins. AlphaFold, a revolutionary new AI tool holds the potential to give us the power to evaluate protein structures like never before, further breaking barriers for advancements in research as well as medicine.
Proteins are nutrients essential to build muscle, but more credit is due to these complex, intricate macromolecules that represent the core machinery behind almost all biological processes. These molecules are extremely versatile in their functionality and this is achieved through the diverse range of possible structures they can form. The folding pattern that proteins undergo after starting off as just a sequence of amino acids is the key factor behind their structural diversity.
AlphaFold, a revolutionary new AI system designed by the company DeepMind opened the door to study proteins like never before by allowing for accurate and efficient prediction of protein structures.
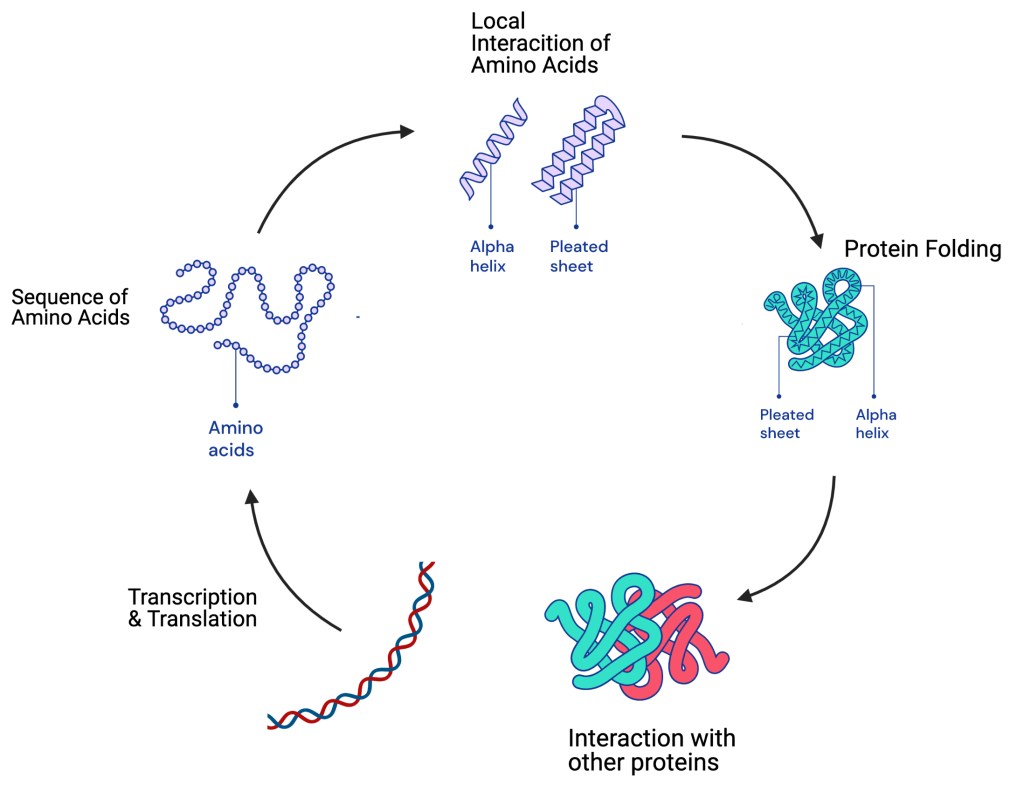
Overview of the biogenesis of a protein. Alpha helices and pleated sheets are so-called secondary structures that are frequently found in proteins.
Why predict protein structures?
There exists a wide variety of proteins in our world, such as transporter proteins (for transportation of molecules in the body), antibodies (for immunity), enzymes (for catalysis), etc. All such protein functions are largely determined by their structure, which is why there is a need to understand protein structures better.
To put this problem further into perspective, consider the viral variants for SARS-CoV-2, the virus which sparked the COVID-19 pandemic. Viral variants found in the UK and Brazil were deemed to have higher transmissibility than the ‘original’ virus and it was understood that the critical factor between these variants was the structure of the spike proteins found on the virus. Discovery of these spike protein structures was the key factor in vaccine design to combat this virus. This clearly illustrates how efficient decoding of protein structures can be a driving force for significant advancements in medicine.
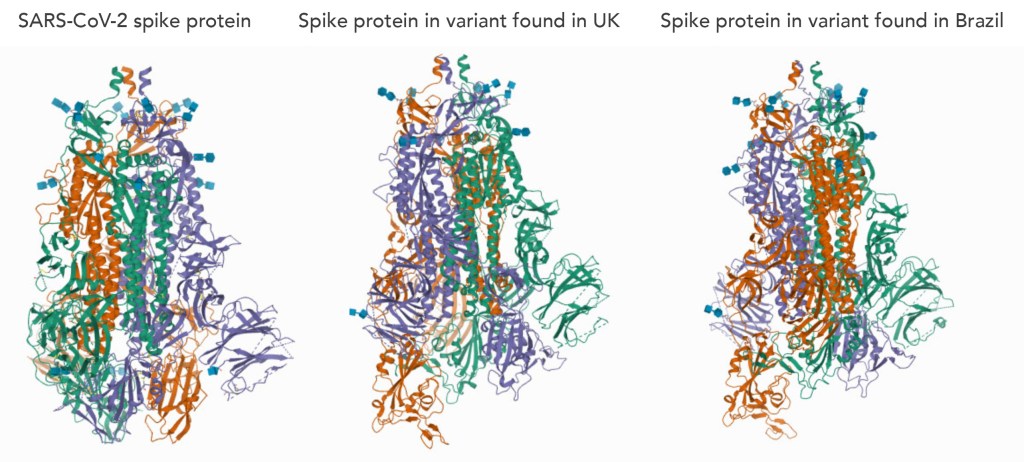
Spike proteins of viral variants shown alongside spike protein found in the ‘original’ SARS-CoV-2 virus (left).
These ideas have led Biologists to ask a crucial question over decades of work, i.e., “How to accurately AND efficiently predict the 3D structure of a protein?”
Enter AlphaFold, the ground-breaking AI system that has broken barriers for advancements in protein structure prediction.
Why AlphaFold?
DeepMind, a general AI company, introduced AlphaFold through their paper Improved Protein structure prediction using Potentials from Deep Learning. AlphaFold highlights significant advancements in the field of protein structure prediction; but what makes AlphaFold so special?
AlphaFold allows for prediction of the 3D structure of a protein solely from its amino acid sequence.
Until now, several experimental techniques (such as X-ray crystallography, cryo-EM and NMR) and non-experimental techniques (heavily based on template-based modeling, i.e., prediction of protein structures by alignment to known/solved protein structures) have been used to successfully deduce the structure of many proteins but there are some caveats to all these approaches: they are either inefficient, expensive, or limited in their ability to decode structures of novel proteins.
AlphaFold has opened the door to efficiently modeling 3D structures of novel proteins in a non-experimental setting, an idea deemed as a “fairy tale” before its advent. This is because there exists an astoundingly large number of possible structural configurations for a given protein (about 10300 for a typical protein!) which made it too challenging a problem to solve without AI. AlphaFold promises to grant scientists the long-sought power to model novel protein structures with great accuracy. Such power to predict the structures of these complex macromolecules simply from their amino acid sequence with increased speed and efficiency is the dream of many biologists.
Furthermore, AlphaFold demonstrated its effectiveness by its commendable achievements in the CASP competition. The CASP competition is a biennial assessment where participating teams are asked to predict structures of previously unseen proteins. In the CASP13 competition (2018), AlphaFold emerged as the best performer!
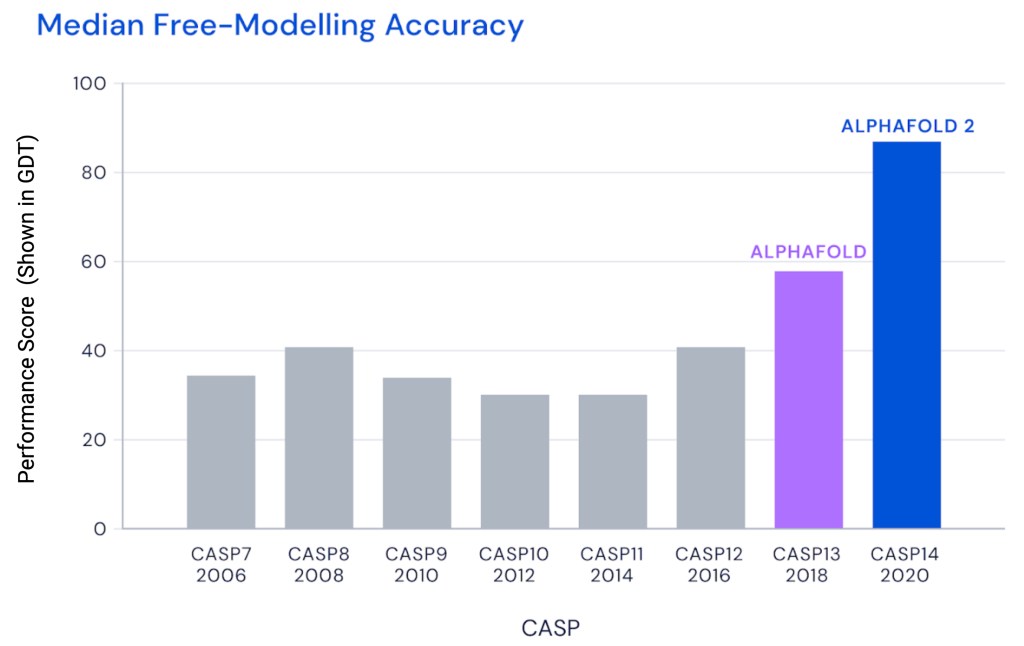
Performance of AlphaFold (Purple) and AlphaFold 2 (Blue) in the CASP competition. GDT (global distance test, ranging from 0 to 100) is the primary metric employed by CASP to quantify accuracy of protein structures.
AlphaFold’s achievements were further surpassed by its successor, AlphaFold II (further discussed below), in the CASP14 competition (2019).
Why AI?
Artificial Intelligence is growing at a tremendous rate and AlphaFold has come forth as one of the most impactful applications of AI. AI systems typically employ large computational architectures known as neural networks. As the name suggests, these systems exhibit “intelligence” and learn patterns and dynamics of a problem simply by being exposed to a vast amount of data relating to the problem.
This core idea of learning is precisely why AlphaFold is able to accurately predict complex structures of proteins despite the vast number of structural possibilities for any given sequence.
At this point, it’s only natural to ask how does AlphaFold achieve this?
Like most AI systems, AlphaFold learns patterns and relationships between different protein structures and sequences by being exposed to a large amount of known protein structures obtained from the Protein Data Bank (PDB). Upon completion of learning, AlphaFold can then be used to predict structures of novel proteins. Specifically, AlphaFold predicts the torsion angles and the distances between amino acid residues in a protein; it can be understood that the torsion angles and residual distances of a protein completely characterize its 3D structure. Therefore, accurate prediction of these angles and distances allows for accurate prediction of protein structure.

What’s next?
Since the release of the AlphaFold I paper in Early 2020, DeepMind has unveiled its sequel AlphaFold II. The improvements of AlphaFold II include some computational and architectural modifications to the original system’s neural network, which in turn leads to more accurate and efficient protein structure prediction. This newer, faster and better version of AlphaFold performed significantly better than its predecessor in the CASP14 competition (90% vs 60%, see figure above).
However, the problem is not 100% solved. Like any other model, there is always room for improvement to learn even more intricate features and to further address ideas such as how proteins fold.
That being said, AlphaFold truly promises to be one of the most significant scientific breakthroughs of our generation. The power to deduce structures of novel proteins with the speed and efficiency that AlphaFold brings to the table clears the pathway for revolutionary breakthroughs in science and medicine.
Learn more about AlphaFold:
- Original article describing AlphaFold I in Nature (2020)
- Original article describing AlphaFold II in Nature (2021)
- Blog post by DeepMind about AlphaFold I
- Blog post by DeepMind about AlphaFold II
- AlphaFold: The making of a scientific breakthrough (YouTube)
About the Author
This post was written by Sarvagya Agrawal. He is an undergraduate student at
University of Toronto looking forward to graduating with a degree in Data
Science/Machine Learning and Molecular Biology. He is heavily interested in Deep
Learning and aims to create efficient models to solve real world problems under the light of both industry and research. Always being fascinated by the field of AI and genomics, he plans to bring some sci-fi theories to life!

This is great! What I don’t get is that AlphaFold is trained on existing models. Existing models are biased, as they really only include proteins that can be crystalized and those that are similar. So then isn’t the AI biased towards predicting structures (or angles and distances) that mirror those seen in proteins that crystalize easily? Can it really predict a *novel* structure?
LikeLiked by 3 people
Thank you for your kind words Dr. Mott.
To answer your question, even though AlphaFold utilizes pre-existing models such as HHblits [for Multiple Sequence Alignment (MSA)], they further employ well known ML techniques such as dropouts (for MSAs), other data augmentation techniques, utilization of auxiliary losses, etc. to make their model more robust to previously unseen structures. These techniques can allow to tune out the bias for early crystallizing structures. The idea here being that AlphaFold doesnt solely rely on the pre-existing models, but rather just uses the insights it can give to improve itself. The network comes up with a probability distribution [P(distance(i,j)|S, MSA(S))] for distances between 2 amino acid residues, i and j given the amino acid sequence S, and the MSA for the sequence, i.e., MSA(S). This probability distribution gives idea about the most likely distances between residues for given sequence, and further helps to model the structure.
That being said, as with all ML/DL models, there is some aspect of uncertainty involved which can be attributed to a number of factors including properties of the datasets as well as the preexisting models used. However, AlphaFold does a good job at modelling the uncertainty as they also include a ‘confidence measure’ (taken care of by another neural network) for their predicted structures, which, as the name suggests, gives an idea about how confident the model is about its prediction. For these reasons, I believe AlphaFold does a fairly good and relatively better job than its competition at predicting novel structures because of the depth of the model and the complexity with which it is trained.
Recently released AlphaFold 2, which employs Transformers as its neural network, does an even better job at ruling out these uncertainties and shows even stronger novel protein structure prediction!
LikeLiked by 1 person
Very interesting Sarvagya! To follow up on Dr Mott’s question, it would be interesting to see the predictions of AlphaFold 1 or AlphaFold 2 on intrinsically disordered domains. I wonder if the researchers can make the difference between “the algorithm does not really know what structure is in this domain” from “this domain does not have a defined structure at all”.
The part that AlphaFold 2 uses Transformers is very interesting as well. There seems to be even more difference between AlphaFold2 and AlphaFold 1 than between AlphaFold 1 and earlier methods. Do the authors claim that the quantum leap is due to Transformers?
For those who want to know more about Transformers, you can have a look at this (technical but) nicely explained blog post.
https://jalammar.github.io/illustrated-transformer/
LikeLiked by 1 person
Thanks for the explanation Sarvagya, I will look into Transformers some more!
LikeLiked by 1 person
Enjoyed reading your post Sarvagya! Really well written, and I think it’s important that ideas like this are communicated in ways that are easy to understand regardless of one’s discipline, which you’ve done! Also, good luck with your goals, I know you can reach them!
LikeLiked by 2 people