
Genetic disorders are hard to cure because they are caused by a defect in the genes of the patient. Gene therapy promises to give patients a second chance by changing their genes. Recently, a new gene-replacement therapy was approved for the Spinal Muscular Atrophy 1, but what does it mean and how does it work?
Gene therapy is about manipulating genes. As weird as that sounds, it can be advantageous if implemented ethically towards a noble goal, such as curing a disease. One of these diseases for which a treatment by gene therapy has been approved by the U.S. Food and Drug Administration (FDA) is Spinal Muscular Atrophy 1 (SMA1).
SMA1 is a motor neuron disease that occurs in 1 out of 10,000 births every year, and it is responsible for the highest number of infant deaths caused by a genetic disease. It is the most severe subtype out of the four subtypes of SMA. SMA1 affects the patients at early ages where symptoms occur within the first six months, leading to the inability to sit unsupported and the need for ventilation support. Sadly, most previous SMA1 patients died before reaching the age of 2. Therefore, it is important to find a solution for this disease, and this is where gene-replacement therapy steps in. It involves the overriding or the replacement of a faulty gene with a functional one. This gene therapy led to striking improvements in patients’ motor functions and achievements of milestones that other SMA1 patients did not achieve.
Long story short, the disease is caused by having low amounts of the survival motor neuron SMN protein. The SMN protein can be produced by two genes, SMN1 and SMN2. The former is the one that produces full-length functional protein. The latter produces mostly a shorter protein, where exon 7 is spliced out; it also produces a low amount of full-length SMN protein. Therefore, retaining the two copies of SMN2 and losing both copies of SMN1 results in a 97% risk of developing SMA1. So you can now imagine what would be the cure for this disease: to get more functional SMN protein in the patient’s body. This might sound very simple, but what is crucial to know is that time matters! The earlier this protein is available for the affected infants, the better, as they are in a life stage where significant development of the body and motor functions occurs.
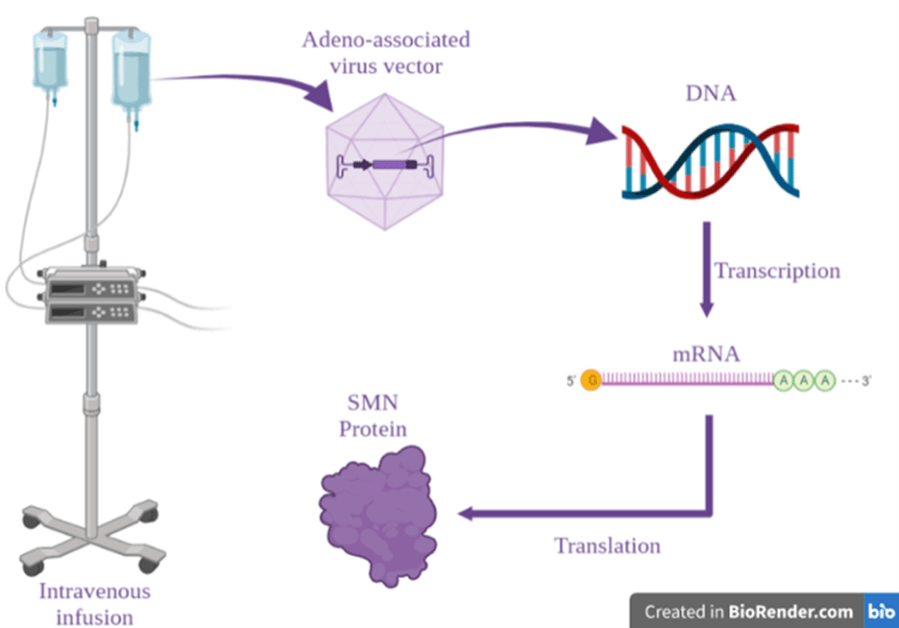
Illustration of the Spinal Muscular Atrophy 1 (SMA1) gene therapy. The adeno-associated virus contains the DNA sequence that codes for survival motor neuron (SMN) protein, which is scarce in SMA1 patients. The virus is transferred directly into the veins of SMA1 patients by intravenous infusion. The DNA sequence is introduced into the cells of the patients, transcribed to produce a messenger RNA and translated to produce the full-length SMN protein. This leads to an increase in the levels of the protein in the patients.
Mendel et al. (2017) developed a gene therapy for SMA1 patients; its mechanism is illustrated in the figure above. The researchers injected the infants that were diagnosed with SMA1 with an adeno-associated viral vector containing a gene that codes for a full-length SMN protein. This gene was then transcribed to mRNA, which was translated to SMN protein in the cells of the infants. In detail, 15 patients were enrolled in this study, where three of them got low doses of this gene-replacement therapy, while the other 12 received higher doses. The results were breathtaking: infants were able to develop motor functions and achieve milestones that previous SMA1 patients, who did not receive gene therapy, could not achieve. For example, some patients were able to sit unsupported and roll over. Additionally, all the patients did not need permanent ventilation at the age of 20 months, where historically only 8% of the patients could achieve that!
The researchers scored the motor skills and functions of the patients, such as their limb movements, on a scale from 0 to 64. To see if the gene-replacement therapy was effective, the researchers compared the motor function score of the infants with a score that was never reached by previous SMA1 patients, which is 40 (see figure below). Amazingly, 11 out of 12 patients, who received the high dose of the gene therapy, had improved motor functions over time and were able to maintain a score above 40.
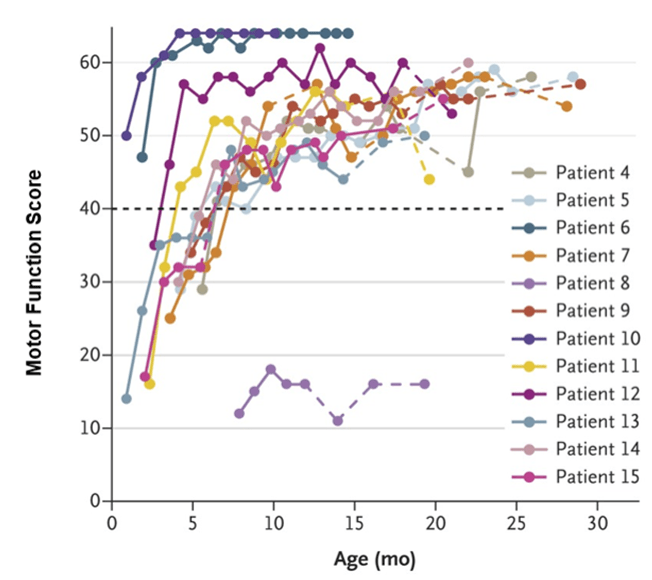
Results of the Spinal Muscular Atrophy 1 (SMA1) gene therapy. 11 out of 12 SMA1 patients who received the high-dose gene therapy had increasing motor functions (movements). In turn, this has led to maintaining a motor function score that is higher than the black dashed line (40). This dashed line shows the score that was never reached by past SMA1 patients. It is most likely that patient 8 did not improve in the motor score as other patients did due to the older age at which this patient received the treatment. The dashed lines of patients’ data are partial or missed assessments that were not considered in the analyses.
Remember that time is key for this gene therapy. Patient 8 was not able to improve and achieve high motor function scores, and when asked about the potential reasons, Dr. W. David Arnold, one of the authors of the study, stated: “It’s hard to know for sure, but it is likely the older age at timing of treatment.” When I met with him virtually, he emphasized that patients who received the therapy at a younger age did better. And if you are wondering why the researchers did not give the therapy earlier, especially since there are tests to diagnose the disease prenatally, it is because the approval they got to implement this study was to give the gene therapy only to symptomatic patients to achieve safety of the experiment. However, the therapy is now FDA approved, and it can be given to patients who are diagnosed with the disease before symptoms occur. Strikingly, the patients of this study are now eight to nine years old, which is amazing when it comes to SMA1, where patients mostly don’t survive beyond two years old.
As amazing as the results may sound, unfortunately, there were many adverse effects, especially on the liver of the patients who received high doses of the therapy. Therefore, low doses, which result in lower improvements in the motor functions, are applied currently. But… are there any alternatives for SMA1 gene therapy? Yes, there are. According to Dr. Arnold, there are two drugs that work against SMA subtypes, which are Nusinersen and Risdiplam. These drugs affect the mRNA splicing of exon 7 to make a functional SMN protein. However, they need to be taken regularly and not as a one-time injection as gene-replacement therapy.
To conclude, the results of the SMA1 gene therapy provide promises to what a great impact gene therapy can have. However, it is really hard to decide where to stop gene therapy; the borderline for that is unclear. Obviously, it has its own benefits when it comes to curing deadly diseases. But… it would be an unrealistic world (even if it was real) if we are all perfect and similar in how we function or even how we look. Therefore, gene therapy is still a topic of debate and ethical concerns, where many studies for each disease are needed before the therapy is approved and used by patients.
Learn more about SMA1 gene therapy
- Gene Therapy for SMA Type 1: Evelyn’s Story (Youtube video)
- SMA Type 1: How Gene Therapy Works (Youtube video)
About the Author
Tasneem Al Mudarris is a student at UTSC pursuing a specialist in molecular biology and biotechnology. She is passionate about research in the field of genetics and genomics, and she thrives for new experiences every day. In fact, she recently discovered that she loves Drosophila flies from a new research experience 🙂
References
Arnold, Kassar, D., & Kissel, J. T. (2015). Spinal muscular atrophy: Diagnosis and management in a new therapeutic era: Spinal Muscular Atrophy. Muscle & Nerve, 51(2), 157–167. https://doi.org/10.1002/mus.24497
Commissioner, O. of the. (n.d.). FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. Retrieved April 19, 2022, from https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy
Commissioner, O. of the. (n.d.). FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. Retrieved April 21, 2022, from https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease#:~:text=The%20most%20common%20side%20effects,of%20experiencing%20serious%20liver%20injury.
Commissioner, O. of the. (n.d.). FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. Retrieved April 19, 2022, from https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy
Dr. W. David Arnold. The Arnold Lab. (n.d.). Retrieved April 19, 2022, from https://u.osu.edu/arnold.780/dr-arnold/
Gene therapy for SMA type 1: Evelyn’s story – youtube. (n.d.). Retrieved April 19, 2022, from https://www.youtube.com/watch?v=yRrqbvUv6gQ
Mendell, Al-Zaidy, S., Shell, R., Arnold, W. D., Rodino-Klapac, L. R., Prior, T. W., Lowes, L., Alfano, L., Berry, K., Church, K., Kissel, J. T., Nagendran, S., L’Italien, J., Sproule, D. M., Wells, C., Cardenas, J. A., Heitzer, M. D., Kaspar, A., Corcoran, S., … Kaspar, B. K. (2017). Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. The New England Journal of Medicine, 377(18), 1713–1722. https://doi.org/10.1056/NEJMoa1706198
SMA Type 1: How gene therapy works – youtube. (n.d.). Retrieved April 19, 2022, from https://www.youtube.com/watch?v=iBmyXr_o1hU
Werner, C. (2020, February 25). Prenatal tests for spinal muscular atrophy: Types, risks, and more. Healthline. Retrieved April 19, 2022, from https://www.healthline.com/health/spinal-muscular-atrophy/prenatal-genetic-testing

I am so proud of you.
Go ahead and show me more advancement and get the Nobel price.
You are the one.
LikeLiked by 3 people
Nice work Tasneem! Your blog post was very informative … I am happy to hear that you are enjoying your research experiences.
LikeLiked by 1 person
Thank you Kyle! Without your great efforts, this blog post wouldn’t be as good as it is now.
LikeLiked by 1 person
You’re too kind haha… all the best!
LikeLike